




La preparación es clave: breve lista de verificación de cómo poner en conformidad que el IVD MD con el EU IVDR
El IVDR evolucionó a partir de la IVDD para establecer un “marco más sólido, transparente, predecible y sostenible para los IVDs”
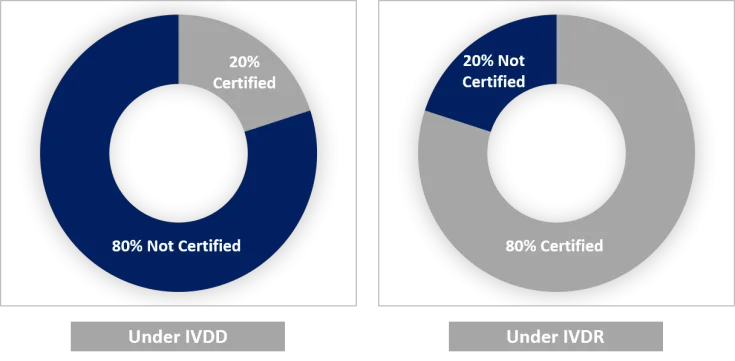
Transición de IVDD a EU IVDR
El tiempo de transición para que los productos sanitarios de diagnóstico in vitro (IVDs) cumplan con el reglamento EU IVD 2016/746 (IVDR) está llegando a su fin: IVDR se implementará por completo a partir del 26 de mayo de 2022. Los nuevos productos sanitarios de diagnóstico in vitro estarán obligados a cumplir con el IVDR, mientras que los IVDs certificados bajo la directiva IVD 98/79/EC (IVDD) no tendrán validez a partir del 27 de mayo de 2024. El tiempo pasa, y se debe tener en cuenta que el cumplimiento del IVDR requiere mucho más trabajo que la IVDD. En este artículo nuestro objetivo es resumir los cambios principales entre la IVDD y el IVDR, incluyendo una guía para que los fabricantes adapten su documentación para el cumplimiento del IVDR.
¿Qué hay nuevo en el IVDR?
El IVDR evolucionó a partir de la IVDD para establecer un “marco más sólido, transparente, predecible y sostenible para los IVDs”. Con esta premisa, el IVDR incorpora los mismos procesos regulatorios básicos de la IVDD y agrega nuevos requerimientos. Algunos de los nuevos requisitos incluyen:
- Definiciones ampliadas y aclaradas para IVDs.
- Nueva clasificación de riesgo: desde clase A (riesgo más bajo) a clase D (riesgo más alto).
- Mayor responsabilidad de los Organismos Notificados (NB) para evaluar si un producto cumple los criterios requeridos.
- Mayores estándares de calidad y seguridad para IVDs.
- Supervisión y trazabilidad mejoradas a lo largo de la cadena de suministro y el ciclo de vida del IVD: aplicación de los identificadores únicos de dispositivos (UDI) y requerimiento de actualizar los datos de fabricación en la Base de datos pública europea para productos sanitarios (EUDAMED).
Definiciones y alcance del IVDR
La definición de IVD se amplía y aclara en el IVDR para incluir más tipos de dispositivos, como servicios de diagnóstico, pruebas genéticas, diagnósticos complementarios y software. También se introducen algunas definiciones nuevas en el IVDR, tales como las pruebas «prueba diagnóstica en el lugar de asistencia al paciente” y las “pruebas diagnósticas para selección terapéutica”. Se debe tener en cuenta que los servicios de prueba que se ofrecen a través de Internet también están bajo el alcance del IVDR cuando se comercializan en la UE.
Nueva clasificación IVD basada en el riesgo
Uno de los cambios más significativos introducidos por el IVDR es la nueva clasificación para los IVDs. El IVDR especifica un conjunto claro de reglas para clasificar los IVD en cuatro categorías de riesgo, que van desde la clase A (riesgo más bajo) a la clase D (riesgo más alto). En particular, los fabricantes deben determinar la clase de IVD independientemente de cómo se clasificó en la IVDD. El Anexo VIII del IVDR proporciona la lista de reglas requeridas para la clasificación de IVD.
Los fabricantes deben tener en cuenta que, según la IVDD, la mayoría de los IVD no requerían supervisión de NB y podían autocertificarse (~ 80%). Por el contrario, el IVDR le da la vuelta a esta relación, y alrededor del 85% de los dispositivos ahora se incluyen en clases de riesgo que requieren la supervisión de los NB.
Mayor responsabilidad de los NBs
Bajo el IVDR, los NBs han adoptado competencias más altas para las evaluaciones científicas y técnicas. En consecuencia, convertirse en Organismo Notificado requiere criterios más estrictos y periodos de evaluación más prolongados (hasta 12 meses). Como resultado, solo hay 5 NBs disponibles en la Unión Europea para el número creciente de IVD que requieren la supervisión de NB: es decir, los NBs son cuellos de botella. Los fabricantes pueden tener en cuenta este hecho al planificar los plazos para colocar un IVD en el mercado de la Unión Europea.
Mayores estándares de calidad y seguridad
El IVDR se centra más en la gestión de todo el ciclo de vida de los IVD, incluida una evaluación continua de las fases posteriores a la comercialización del producto:
- Requisitos para que los fabricantes demuestren sistemas de gestión de calidad (SGC) eficaces
- Los requisitos para demostrar evidencia clínica (denominados “evidencia de desempeño” en el IVDR) son más estrictos y se vuelven progresivamente más estrictos a medida que aumenta la clase de riesgo. Además, la evaluación del desempeño se basa en:
- Validez científica
- Rendimiento analítico
- Rendimiento clínico
- Requisitos novedosos y más altos para el seguimiento del rendimiento posterior a la comercialización para evaluar continuamente el IVD durante todo el ciclo de vida del dispositivo.
UDI y EUDAMED: mayor transparencia y trazabilidad
Una de las principales razones para desarrollar el IVDR fue incrementar la transparencia y trazabilidad de los productos comercializados en el mercado de la UE. Para ello, el IVDR introduce los identificadores únicos de dispositivo (UDI), códigos colocados en los IVD para ayudar en su identificación y trazabilidad. La asignación de UDI es realizada por algunas entidades emisoras y debe renovarse cada cinco años. Para facilitar dicha trazabilidad, los fabricantes son responsables de introducir UDI y otros datos relevantes en la base de datos pública europea (EUDAMED). En consecuencia, EUDAMED ayuda a mejorar la transparencia al funcionar como un sistema de registro, notificación y difusión de información para productos de IVD comercializados en el mercado de la UE.
¿Te sientes listo? AKRN te ayudará
El IVDR proporciona un nuevo marco regulatorio para la colocación de IVDs en el mercado de la UE. El alcance ampliado de la supervisión regulatoria ha pillado desprevenidos a algunos fabricantes, y aquellos que han pospuesto la transición al IVDR pueden sentir la presión de adaptarse pronto. AKRN se especializa en la transición de IVDD a IVDR. Como parte de nuestros servicios regulatorios, AKRN puede ayudar a los fabricantes de IVD a cumplir con los requisitos necesarios para colocar IVD en el mercado de la UE. Dichos servicios, incluida la preparación de la documentación técnica y las evaluaciones de desempeño, ayudarán a los fabricantes a lograr una transición fluida y el cumplimiento de la normativa de la UE.
Por Maria Nyakern, CEO de AKRN Scientific Consulting