




Notificaciones de seguridad para investigaciones clínicas de productos sanitarios bajo el MDR
Las notificaciones de seguridad son un aspecto crucial para el éxito de una investigación clínica de productos sanitarios, los cuales deben cumplir con las normas de Buena Práctica Clínica (BPC) y el reglamento de productos sanitarios EU 2017/745 (MDR)

¿Qué se comunica?
El artículo 80 (2) del MDR enumera los acontecimientos que deben notificarse durante la realización de una investigación clínica:
- Cualquier acontecimiento adverso grave (AAG) con una relación causal con el producto en investigación, el comparador o el procedimiento en investigación o cuando dicha relación causal sea razonablemente posible.
- Cualquier deficiencia del producto que podría haber dado lugar a un AAG si no se hubieran tomado las medidas adecuadas, no se hubiera intervenido o las circunstancias hubieran sido menos afortunadas.
- Cualquier nuevo hallazgo en relación con cualquier evento referido a los puntos anteriores.
Para arrojar más luz sobre la relación causal del evento con el procedimiento o dispositivo, el MDCG 2020-10/1 define cómo clasificar los AAG según cuatro niveles de causalidad:
- No relacionado
- Posible: relación débil, pero que no puede descartarse por completo.
- Probable: relación relevante que no puede ser razonablemente explicada por otra causa.
- Relación causal: relación más allá de una duda razonable.
Por lo tanto, se espera que los patrocinadores informen de cualquier AAG posible, probable y causal relacionado con el producto o procedimiento en investigación.
Obsérvese que cuando el nivel de causalidad es incierto, el promotor no debe excluir la relación y el acontecimiento debe clasificarse como "posible".
Téngase en cuenta que las investigaciones de seguimiento clínico poscomercialización de los productos con marcado CE utilizados dentro del uso previsto cubierto por el marcado CE deben seguir los mecanismos de notificación de vigilancia.
¿A quién se comunica? ¿Cuándo?
Los sucesos notificables deben comunicarse a las Autoridades Nacionales Competentes (ANC). La lista y el contacto de dichas ANC operativas pueden encontrarse en la página web de la Comisión y en el siguiente enlace. Además, los Estados miembros también pueden exigir que se informe por separado a los comités de ética.
En cuanto a los plazos, los investigadores deben informar a los patrocinadores inmediatamente, no más tarde que 3 días naturales después de que el personal del estudio del centro de investigación tenga conocimiento del suceso. El promotor, por su parte, y en función de la categoría del estudio clínico y de la gravedad del acontecimiento adverso, debe seguir los siguientes plazos de notificación:
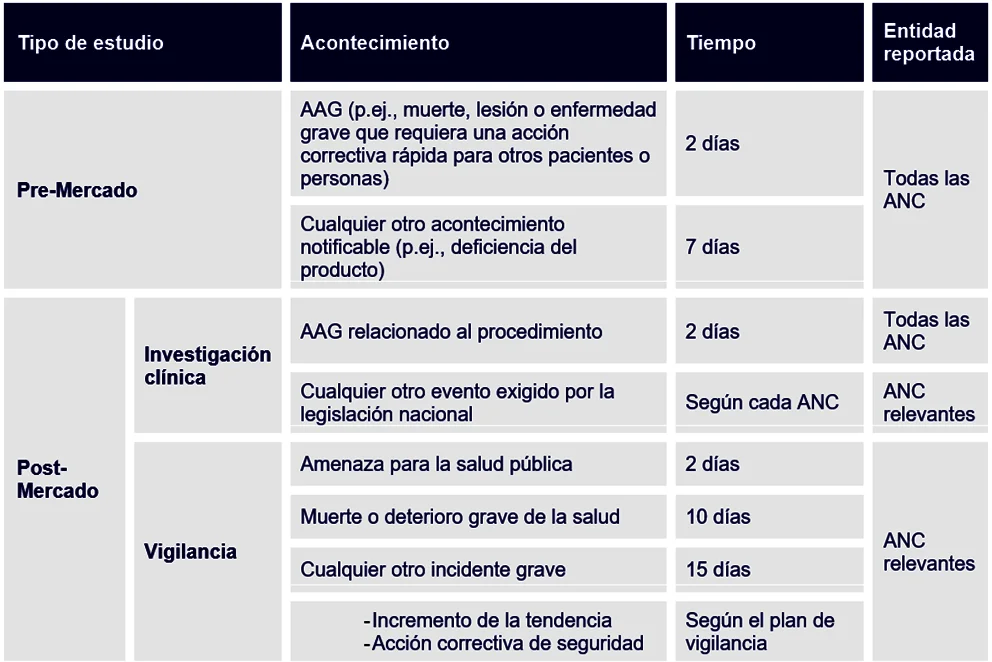
Además, tenga en cuenta que en los estudios observacionales en los que se mantiene la práctica clínical habitual, sólo se notificarán los acontecimientos adversos no esperados que no estén documentados en la información del producto y cuantificados en la documentación técnica, de acuerdo con el plan de vigilancia.
AKRN puede ayudarte
El equipo clínico de AKRN cuenta con una amplia experiencia en el diseño, la realización, el registro y la presentación de informes de investigaciones clínicas, incluidas las notificaciones de seguridad según los requisitos del MDR. Consulte aquí los servicios que ofrecemos.
Además, no dude en ponerse en contacto con nosotros para obtener más información sobre el apoyo que ofrecemos a investigaciones clínicas para dispositivos médicos.
Autor: Albert Negrete Hurtado, PhD, Regulatory Affairs Scientist at AKRN Scientific Consulting